Project 16.2
- PhD student: Ishita Amar
- Supervisor: Joan Barau
- Co-Supervisors: Jan Padeken
- Further TAC-members: Antoine Molaro, France Maxim Greenberg (Institut Jacques Monod)
- Research Group
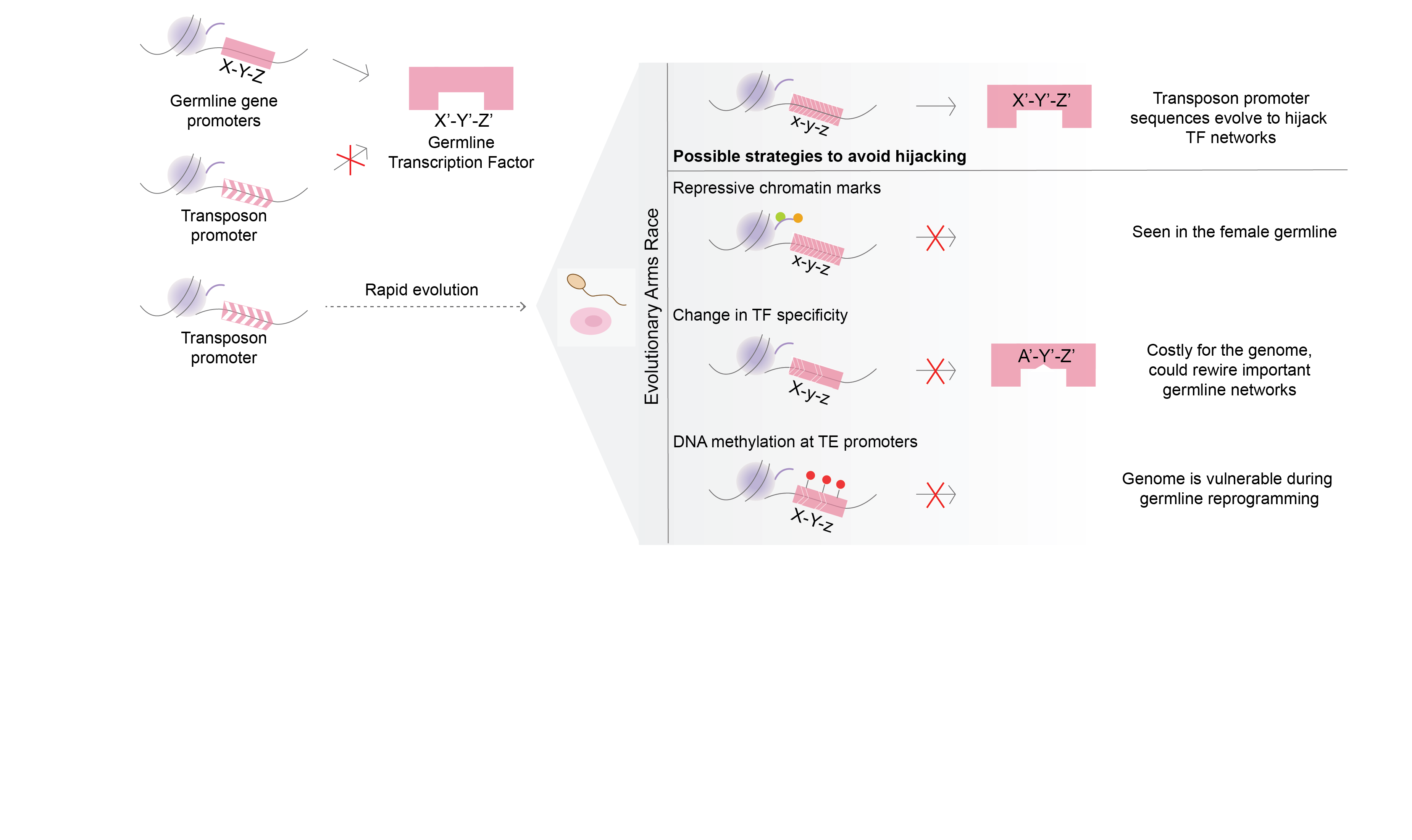
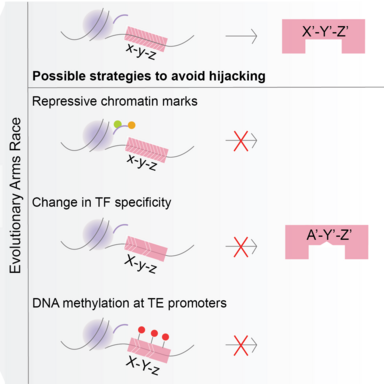
The project focusses on the evolutionary arms-race between transposons and germline transcription factors. Transposons achieve evolutionary success only when they can propagate in the germline and can be inherited in the next generation. One of the strategies to achieve this is to ‘hijack’ germline gene networks by mimicking germline gene promoters. Our aim is to study how this ‘hijacking’ takes place in germ cells.
Transposable elements (TEs) are abundantly found in most eukaryotic genomes. Their evolutionary success relies on their ability to replicate in the germline and be inherited in the next generation. TEs are often repressed by epigenetic mechanisms like DNA methylation. However, during germline epigenetic reprogramming, the genome loses almost all its methylation putting the germline integrity at risk of TE activity. This implies that the copies of TEs that are present in the genome must be those that could effectively hijack germline networks during the stages where methylation is dynamic, but whose activity was not sufficiently detrimental for its alleles to be purged from the population.
In order to study this, we use a DNMT3C-KO model. DNMT3C is a germline-specific DNA methyltransferase that is known to methylate young transposon promoters. It is an evolutionary young DNMT, found only in Muroidea. Mouse mutants with inactive DNMT3C are male infertile, and sperm is not produced due to excessive TE activity and meiotic arrest. The focalized DNA methylation defects of DNMT3C-KO are a perfect background model to study which networks were hijacked by transposons during evolution.
Our aim is to identify the activators of transposons that have been co-opted during evolution. To find these, we will first profile the chromatin marks that are present on transposon promoters in the absence of DNMT3C. Then we will compare the marks with those present on germline genes and see if some chromatin signatures are common. These could give clues into regulators and networks that have been potentially hijacked.
Additionally, we will use comparative genomics and in vitro functional assays to explore how a strong selective pressure to avoid binding to transposons affects the evolution of germline transcriptional networks that are essential for reproduction (e.g. meiosis).