Project 1.3
- PhD student: Cassandra Theresa Mitchell
- Supervisor: Joachim Burger
- Further TAC-members: Christof Niehrs, Jan Padeken
- Research Group
My research project explores major selective processes in human evolution over the past millennia, focusing on their detectable traces in epigenetic modifications. The focus will be on previously identified epigenetic variants in the genomes of prehistoric humans with diverse lifestyles, and the aim is to discover their function as well as their evolutionary advantages in the associated lifestyle.
Methylations are epigenetic modifications and serve as a regulatory factor between the interplay of the genotype and the phenotype. In contrast to genetic modifications, epigenetic modifications are reversible and occur more rapidly. Differential methylation can result from a multitude of factors, such as diet, environment, and stress levels (Alegría-Torres, Baccarelli, & Bollati, 2011).
Palaeogenetics offers a window into the lives of past populations by analysing the genomes of humans who lived centuries or even millennia ago. Using ancient DNA (aDNA) extracted from skeletal remains, researchers reconstruct genomes with the help of advanced bioinformatics tools and population genetic statistical inference.
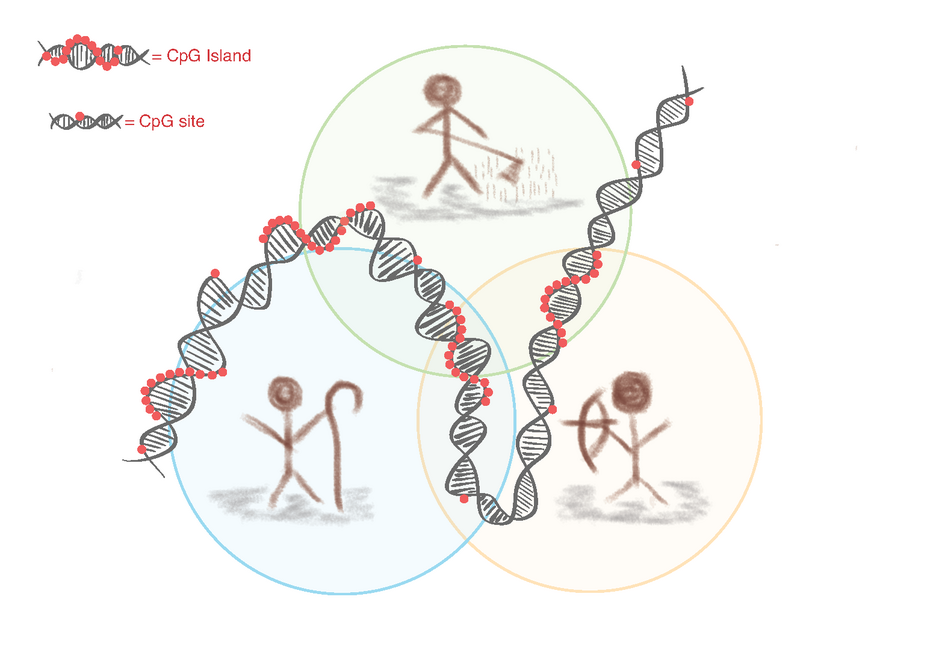
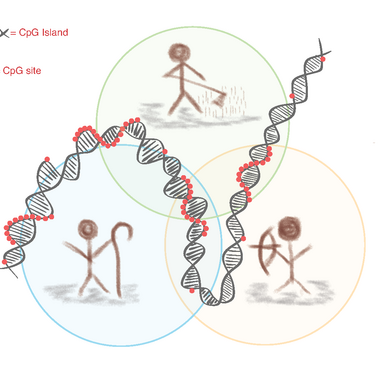
Ancient methylome analysis offers new insights into the lifestyles of past individuals and populations. Unlike the genome, which reflects slow genetic mutations, the methylome also captures rapid epigenetic changes. Of particular interest are regions rich in CpG sites, so-called CpG islands, as these are the primary locations of methylation in humans.
A previous GenEvo PhD project focused on comparing methylation levels between hunter-gatherer and early farming lifestyles. The shift from a foraging to a sedentary, agricultural way of life is considered one of the most profound lifestyle transitions in human history. The technical idea behind this was that post-mortem DNA undergoes specific chemical changes that can be used to infer past methylation patterns. While unmethylated cytosines degrade into uracil over time, methylated cytosines are converted into thymine. This chemical distinction allows researchers to computationally reconstruct ancient methylation profiles. Using this approach, several differentially methylated regions (DMRs) were identified in silico — some of which are believed to have been influenced by natural selection.
In my project, the findings of the previous project will be taken up and further investigated in the lab. As a first step, we aim to verify whether the differentially methylated regions (DMRs) inferred in silico can be confirmed through bisulfite- and Nanopore sequencing in historical genomes. As a next step, I would like to use genome and epigenome editing techniques to investigate the functional effects of different methylation states in cell lines.
Reference: Alegría-Torres, J. A., Baccarelli, A., & Bollati, V. (2011). Epigenetics and Lifestyle. Epigenomics, 3(3), 267–277. doi.org/10.2217/epi.11.22