Project 9.3
- PhD student: Ethan Reece
- Supervisor: Helen May-Simera
- Co-Supervisors: Susanne Foitzik
- Further TAC-members: Jan Padeken , Hans-Peter Wollscheidt (IMB, Mainz)
- Research Group
Bardet-Biedl Syndrome (BBS) is a genetic disorder associated with dysfunctional primary cilia, microtubule-based sensory organelles. While BBS proteins are best known for their ciliary roles, they also localize to other cellular compartments, including the nucleus. These alternative localizations are interesting, as ciliary defects alone do not fully explain BBS pathology. This project investigates the significance and evolution of nuclear BBS protein functions and how these roles may contribute to BBS disease pathology.
Cilia and flagella are microtubule-based organelles that extend into the extracellular space. In eukaryotes, cilia occur in two forms: motile cilia, which generate movement, and immotile primary cilia, which function as sensory and signaling hubs. The proteins required for their assembly, maintenance, and function are highly conserved across species. In humans, mutations in ciliary proteins can disrupt multiple organ systems, resulting in ciliopathies, a group of disorders caused by defective cilia. Bardet-Biedl Syndrome (BBS), often considered the archetypal ciliopathy, is caused by mutations in BBS genes and can result in phenotypes such as polydactyly, retinal degeneration, kidney malformations, and obesity.
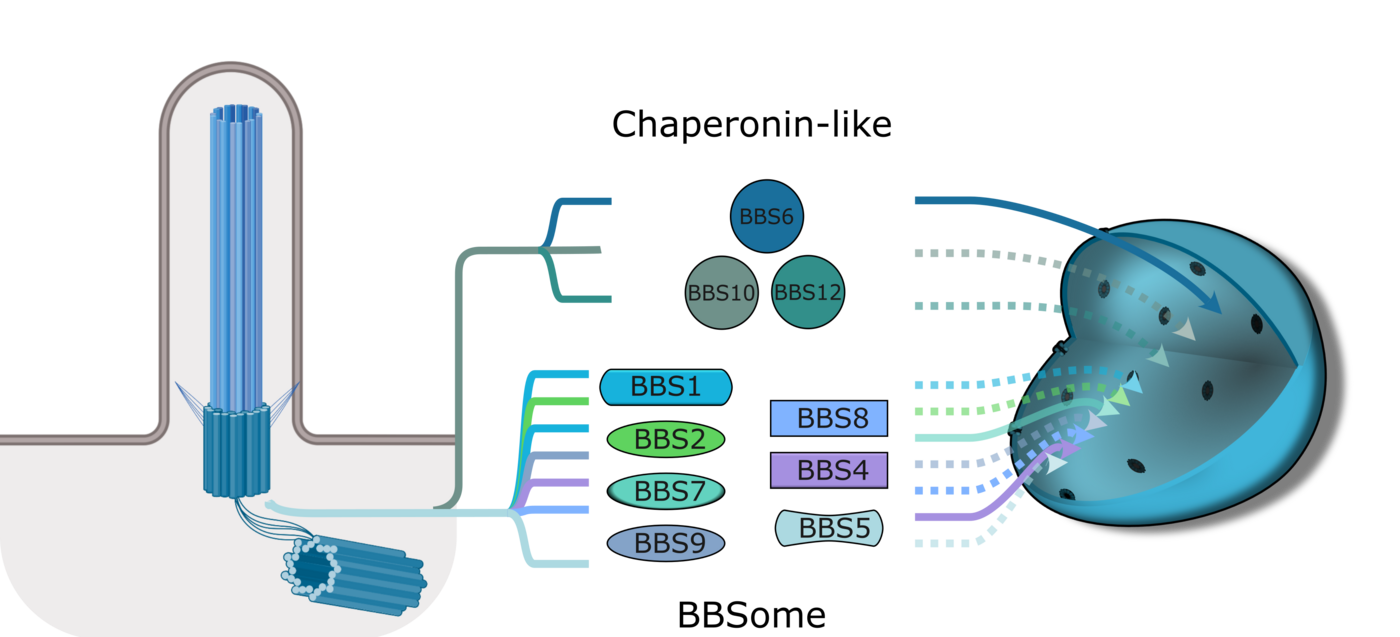
BBS proteins are best known for their roles in ciliary trafficking. Some assemble into the hetero-octameric BBSome complex, which links cargo to the intraflagellar transport (IFT) machinery, while others function as chaperonin-like proteins that assist BBSome assembly. Although initially characterized in the context of cilia, BBS proteins have since been implicated in additional cellular processes, including mitosis, cytokinesis, and the DNA damage response. More recently, several BBS proteins have been shown to localize to the nucleus, suggesting that they may also have nuclear functions. Since ciliary dysfunction alone does not fully explain the complexity of BBS symptoms, investigating these alternative roles is essential for understanding disease mechanisms and improving clinical outcomes.
This project aims to investigate the functional significance of nuclear BBS protein localization, by addressing three complementary questions.
- We will examine whether BBS proteins accumulate in the nucleus under stress conditions in cultured human cells. Previous studies have reported nuclear localization of certain BBS proteins in response to ethanol or etoposide exposure. We will label further BBS proteins and assess their nuclear presence under a broader range of stress conditions using immunofluorescence and Western blotting. We will also explore the impact of nuclear-localized BBS proteins on gene regulation by analyzing transcriptomic changes under these stress-induced overexpression conditions.
- We will assess whether stress-induced nuclear localization of BBS proteins is conserved across phylogenetically distant organisms. Orthologs of BBS proteins in diverse eukaryotes, including Chlamydomonas reinhardtii, Caenorhabditis elegans, and Danio rerio, contain predicted nuclear localization or export signals. Interestingly, these signals do not follow patterns of mitotic inheritance, suggesting that nuclear localization arose independently in different lineages. Based on these predictions and experimental feasibility, we will establish selected model systems and assess whether stress conditions similarly influence nuclear localization of BBS homologs.
- We will investigate the nuclear functions of BBS proteins by identifying their interaction partners. We are developing an in vivo proximity-dependent biotinylation approach, expressing BBS proteins fused to a biotin ligase and nuclear localization sequences. After biotin incubation, biotinylated proteins will be isolated and identified via mass spectrometry to generate nuclear interactomes.
Together, these approaches will clarify the significance of BBS protein localization to the nucleus and broaden our understanding of how non-ciliary roles contribute to Bardet-Biedl Syndrome pathology.